


I'm a bit skeptical of AlphaFold 3

So this happened: DeepMind (with 48 authors, including a new member of the British nobility) decided to compete with me. Or rather, with some of my work from 10+ years ago.
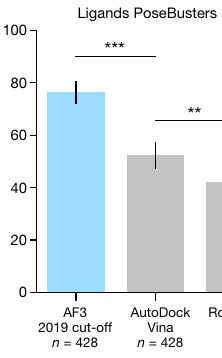
Apparently, AlphaFold 3 can now predict how a given drug-like molecule will bind to its target protein. And it does so better than AutoDock Vina (the most cited molecular docking program, which I built at Scripps Research):
On top of this, it doesn’t even need a 3D structure of the target. It predicts it too!
But I’m a bit skeptical. I’ll try to explain why.
Consider a hypothetical scientific dataset where all data is duplicated: Perhaps the scientists had trust issues and tried to check each others’ work. Suppose you split this data randomly into training and test subsets at a ratio of 3-to-1, as is often done:

Now, if all your “learning” algorithm does is memorize the training data, it will be very easy for it to do well on 75% of the test data, because 75% of the test data will have copies in the training data.
Scientists mistrusting each other are only one source of data redundancy, by the way. Different proteins can also be related to each other. Even when the sequence similarity between two proteins is low, because of evolutionary pressures, this similarity tends to be concentrated where it matters, which is the binding site.
Lastly, scientists typically don’t just take random proteins and random drug-like molecules, and try to determine their combined structures. Oftentimes, they take baby steps, choosing to study drug-like molecules similar to the ones already discovered for the same or related targets.
So there can be lots of redundancy and near-redundancy in the public 3D data of drug-like molecules and proteins bound together.
Long ago, when I was a PhD student at Columbia, I trained a neural network to predict protein flexibility. The dataset I had was tiny, but it had interrelated proteins already:

With a larger dataset, due to the Birthday Paradox, the interrelatedness would have probably been a much bigger concern.
Back then, I decided that using a random train-test split would have been wrong. So I made sure that related proteins were never in both “train” and “test” subsets at the same time. With my model, I was essentially saying “Give me a protein, and (even) if it’s unrelated to the ones in my training data, I can predict …”
The authors don’t seem to do that. Their analysis reports that most of the proteins in the test dataset had kin in the training dataset with sequence identity in the 95-100 range. Some had sequence identity below 30, but I wonder if this should really be called “low”:

This makes it hard to interpret. Maybe the results tell us something about the model’s ability to learn how molecules interact. Or maybe they tell us something about the redundancy of 3D data that people tend to deposit? Or some combination?
Docking software is used to scan millions and billions of drug-like molecules looking for new potential binders. So it needs to be able to generalize, rather than just memorize.
But the following bit makes me really uneasy. The authors say:
The second class of stereochemical violations is a tendency of the model to occasionally produce overlapping (clashing) atoms in the predictions. This sometimes manifests as extreme violations in homomers in which entire chains have been observed to overlap (Fig. 5e).
If AlphaFold 3 is actually learning any non-obvious insights from data, about how molecules interact, why is it missing possibly the most obvious one of them all, which is that interpenetrating atoms are bad?
On the other hand, if most of what it does is memorize and regurgitate data (when it can), this would explain such failures coupled with seemingly spectacular performance.
I agree with the skepticism of the author. Due to the lack of clarity on training/test bias avoidance and evaluation methods, it's not yet clear whether alphafold 3 performance in protein-ligand interaction are conclusive.
I have seen this first hand when working with CHEMBL data, a naive data-split will cause you to overestimate your model performance as (1) assay results for a particular ligand can be obtained from same/different experiments which leads to data leakage (2) many assays test for ligands with similar molecular structures, and these similar structures can end up in different train/test datasets.
One thing I would also like to see the alphafold team address is model generalization. Here is a good experiment:
1. Take your train and test ligands.
2. Compute the mean normalized jaccard index between each test ligand and train ligands.
3. Bucketize the test ligands for every 10th percentile of the normalized Jaccard Index difference.
4. Report the performance comparing alpha fold and vina.
In other words, to test model generalizability, we want to see the extent to which ligands have to be similar to training before we see gains with respect to simulation methods (e.g. vina). From experience, I expect to see some signals of overfitting.
Also, thank you for your work on vina, I learned quite a lot about this space just from reading your repo code!